materialyzeai/megnet
A graph neural net for crystals that actually shipped
MEGNet applies DeepMind's graph networks to predict material properties from molecular structures—now frozen in TensorFlow amber while the team moved to PyTorch.
Not currently ranked — collecting fresh signals.
star history
What it does
MEGNet (MatErials Graph Network) trains graph neural networks to predict properties of molecules and crystals directly from their structures. Feed it a crystal structure or a SMILES string, and it returns formation energies, band gaps, bulk moduli, or a dozen QM9 molecular properties. The project includes pre-trained models serialized as HDF5+JSON files that load with a single from_file call.
The interesting bit
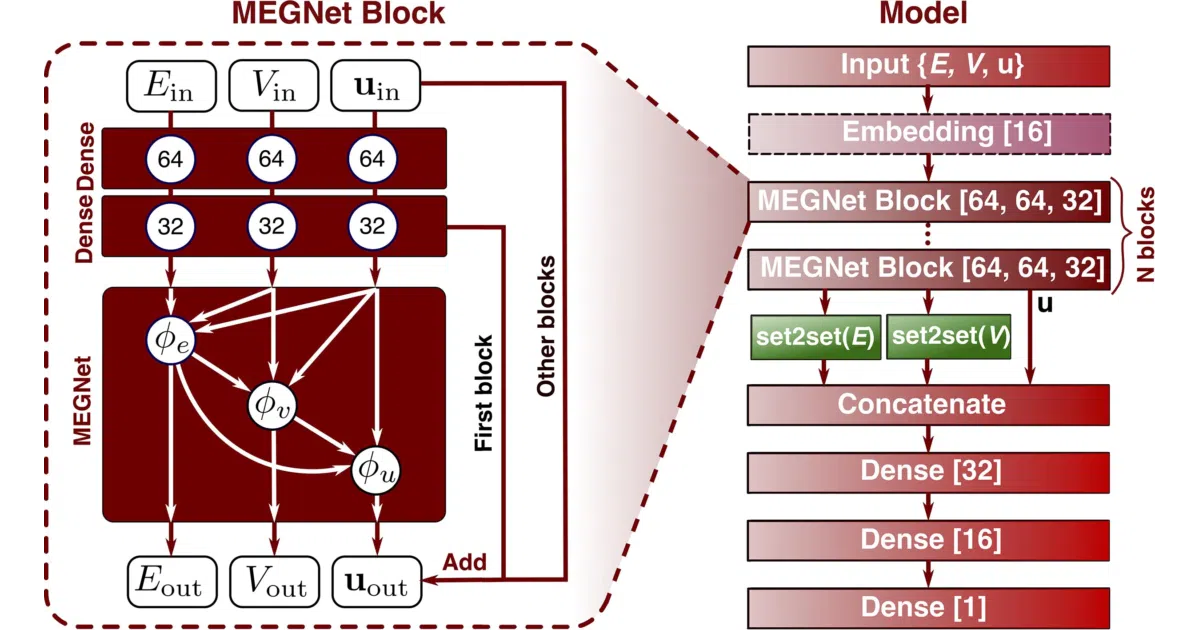
The architecture stacks “MEGNet blocks”—each block prepends two dense layers to a graph network update—allowing the model to tune how far information propagates through the structure. A set2set readout collapses the final graph to a scalar. The authors also found that element embeddings learned from formation energy models transfer well to scarcer properties like elastic constants, which is the kind of practical insight that separates materials ML from generic graph benchmarks.
Key highlights
- Pre-trained models available for QM9 (13 molecular properties) and Materials Project (formation energy, band gap, bulk/shear moduli)
- Published MAEs: 0.028 eV/atom formation energy, 0.33 eV band gap on MP-2018.6.1
- Supports training from
pymatgenStructureorMoleculeobjects with aCrystalGraphconverter - Includes multi-fidelity training examples in a dedicated
multifidelity/folder - Elemental embeddings exposed for transfer learning to smaller datasets
Caveats
- Deprecated: the authors explicitly state this repo is frozen; active development moved to matgl (PyTorch + DGL)
- TensorFlow/Keras implementation only; the README nudges new users toward the DGL reimplementation instead
- QM9 molecular models are not recommended for molecules outside the training set due to limited coverage
- Some structures with isolated atoms will crash
train(); you must filter to valid graphs manually viatrain_from_graphs
Verdict
Worth a look if you’re reproducing the 2019–2020 papers or need a quick baseline on pymatgen structures without rewriting data pipelines. Skip it if you’re starting fresh—follow the deprecation notice to matgl instead.
Frequently asked
- What is materialyzeai/megnet?
- MEGNet applies DeepMind's graph networks to predict material properties from molecular structures—now frozen in TensorFlow amber while the team moved to PyTorch.
- Is megnet open source?
- Yes — materialyzeai/megnet is open source, released under the BSD-3-Clause license.
- What language is megnet written in?
- materialyzeai/megnet is primarily written in Jupyter Notebook.
- How popular is megnet?
- materialyzeai/megnet has 559 stars on GitHub.
- Where can I find megnet?
- materialyzeai/megnet is on GitHub at https://github.com/materialyzeai/megnet.